
Structure-Based Confirmation of Small Molecules in LC/MS Datasets: Application to Metabolite Identification
David Stranz, Scott Campbell, and George Maydwell, Sierra Analytics, Inc., Modesto, CA, 95356 USA
[This publication was originally presented as a poster at CoSMoS 2004.]
Abstract
We present new software, which, based on chemical structure and minimal analyst input, can quickly and accurately identify small molecules in LC/MS-MS/MS datasets.
We illustrate with an application to metabolite ID.
Introduction
Many small-molecule characterization problems can be stated as:
- The sample contains one or more molecules of interest
- A set of candidate structures has been proposed
- Experimental LC/MS and/or LC/MS/MS data have been acquired
- Which, if any, of the proposed molecules are present, and to what extent?
Solution, in most cases, requires time and expert interpretation by the analyst.
In particular, metabolite identification needs two sets of expertise: an expert to propose expected metabolites, and another to determine those actually in the sample. Software present on the MS data systems provides limited help in either area, with most giving only clues to the identity of the metabolite and no actual structures.
In this work, we described the use of in-silico metabolite prediction software with automated interpretation of LC/MS-MS/MS data.
Experimental
Imipramine was incubated with rat liver S9 microsomes. After 1 hour, a sample was withdrawn and analyzed using a data-directed LC / ESI+ / MS / MS2 / MS3 protocol on an LCQ ion trap instrument.
In-silico Metabolite Prediction
The PredictIT Metabolism software from Bio-Rad Laboratories, Inc. was used to predict all potential phase 1 and 2 metabolites for imipramine.
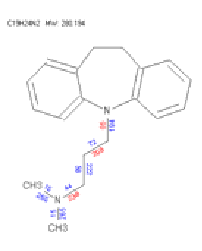
In all, some 43 unique structures were predicted, of which 15 were isomeric. These plus the substrate structure were exported in the form of an SDFile.
Data Analysis
The SDFile containing predicted metabolites and the raw LC/MS dataset were imported into Sierra’s Apex software. MS and MS/MS spectra were
peak detected and centroided.
For each of the proposed structures, the chemical formula was computed and used to generate an isotope cluster spectrum. The structures were used to predict MS/MS fragmentation “spectra” by cleaving all non-cyclic single bonds.
To score the candidate structures against the data, a set of three scoring “chromatograms” was generated for each structure, by computing these metrics for each spectrum in the dataset:
- Similarity (isotope cluster vs. spectrum)
- Isotopic purity (% of spectrum due to molecular ion cluster)
- MS/MS similarity
Scores were normalized to 1.0 within each “chromatogram”.
A total correlation “chromatogram” was computed from the product of the three scoring chromatograms at each point. The overall correlation score for the structure was the maximum in this “chromatogram”. Structures were then ranked by overall correlation score.
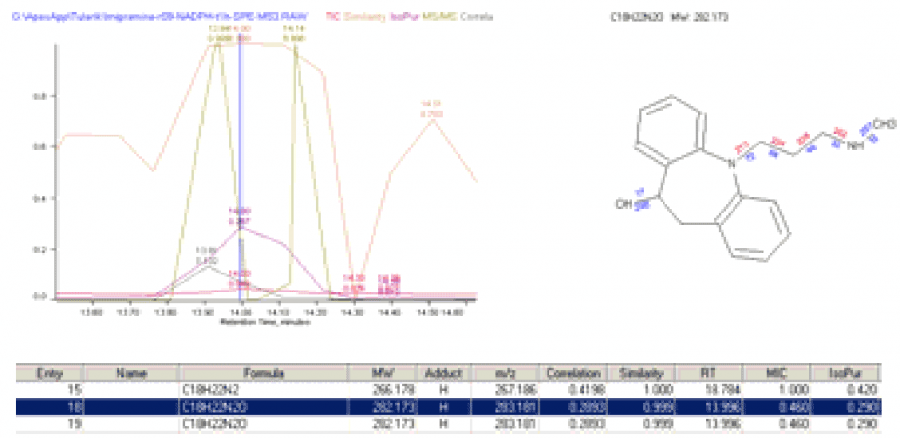
Results
Of the 44 structures (substrate plus 43 unique metabolites), good quality correlation scores were obtained for 11, including imipramine itself.
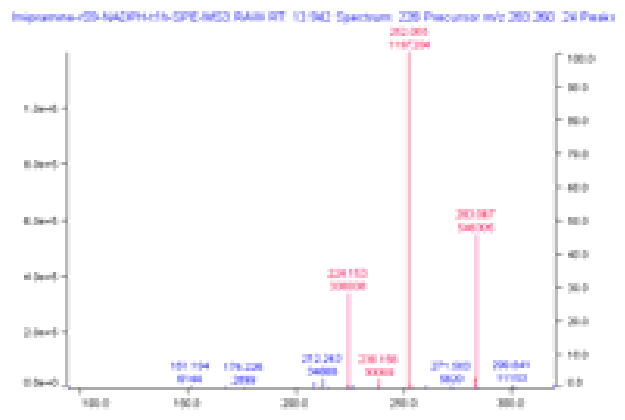
The TIC showed weak features for most of these structures. However, as shown in the figure above, the scoring chromatograms are strongly peaked.
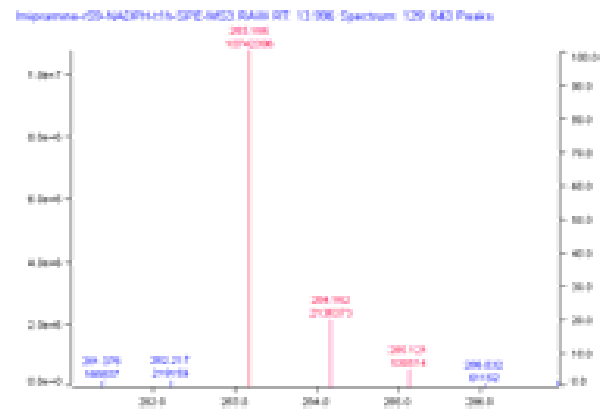
The MS spectrum at this retention time is shown above, with matches between actual and predicted isotopic peaks colored. The match between the corresponding experimental and predicted MS/MS fragmentation is shown below. In both cases, the major features in the spectra are in good agreement with prediction.
Conclusions
In-silico prediction provides structures for the most likely metabolites. In combination with data-directed MS-MS/MS acquisition and automated structure-based analysis of the experimental spectra, the metabolites that actually occur from among those predicted can be readily and confidently identified.
Acknowledgements
Shichang Miao for the use of his imipramine dataset.
Bio-Rad Laboratories, Inc. for the use of Know-It-All® PredictIT Metabolism.