
Identification of In-Vitro Metabolites of Indinavir using Automated LC/MS/MS Acquisition, In-Silico Prediction, and Structure-Based Data Analysis
Casey Hao, Applied Biosystems, Foster City, CA 94404 USA
Scott Campbell and David Stranz, Sierra Analytics
Nicole McSweeney, LHASA Limited, University of Leeds, Leeds LS2 9JT UK
[This publication was originally presented as a poster at ASMS 2004.]
ABSTRACT
Modern hybrid tandem MS instruments can automatically and routinely acquire enhanced and highly sensitive parent and product ion scans for all significant components in an LC eluent stream. For most scientists, prediction of potential xenobiotic metabolites, data processing, characterization of the actual metabolites, and reporting are severe bottlenecks in high-throughput metabolism studies. Lack of reliable and knowledgeable software tools for structural data interpretation is a major contributor to turn-around time.
This work describes a method that couples software for xenobiotic metabolite prediction, automated LC/MS and LC/MS/MS data acquisition, and software for structure-based small molecule identification and characterization to predict, detect, and identify in-vitro metabolites of indinavir.
INTRODUCTION
Using the linear ion trap mass spectrometer to perform enhanced MS survey and product ion MS/MS scans, six rat S9 metabolites of indinavir were identified with rich fragment information. Three of the six metabolites, formed by N-dealkylation and N-oxidation of the aromatic heterocycle and by hydroxylation did not produce the same major fragments observed in the product ion spectrum of indinavir. Three others, an amide reduction and two aromatic and hetero-aromatic ring hydroxylation metabolites, consisted not only of higher mass fragments, but a significant fraction of the lower mass fragments also observed in the product ion spectrum of indinavir.
Identification of the six Phase I metabolites was originally carried out by review and examination of the chromatogram and its associated MS and MS/MS spectra in comparison to the published data from an experienced chemist. The published analysis was made without the assistance of automated metabolite prediction or spectral characterization software.
Using METEOR’s base of more than 300 biotransformation rules, 102 potential indinavir metabolites were predicted. These included the five of the six metabolites previously identified, along with products arising from sulphation, glucuronidation, hydrolysis and other pathways.
The Apex software automatically processed the experimental LC/MS and MS/MS IDA dataset. The metabolite structures predicted by METEOR were used to generate a set of predicted parent ion isotope patterns and product ion fragmentation spectra. By correlation of the predicted and experimental spectra, Apex determined that the most probable predicted metabolites were the same five as previously identified and reported retentions and relative concentrations consistent with the manual analysis. Rejected structures were either inconsistent with observed parent ions or had poor correlation between predicted and observed fragmentation spectra.
EXPERIMENTS AND METHODS
Q-TRAP IDA
Indinavir was incubated with rat liver S9 fractions. Aliquots (10 ?L) were separated by online LC and analyzed with an Applied Biosystems hybrid triple quadrupole / linear ion trap mass spectrometer in Information Dependent Acquisition (IDA) mode, consisting of an enhanced MS survey scan followed by enhanced product ion scans for the two most intense parent ions as dependent MS/MS experiments.
METEOR METABOLITE PREDICTION
METEOR Version 7, from LHASA Limited, is a software system for the prediction of xenobiotic metabolism. It uses a knowledge base of 302 structure-metabolism relationships and justifies its predictions with literature references and examples. It has the capability to reason about factors, which influence metabolism, such as physico-chemical properties, competition with other reactions, or whether the metabolite was administered or generated. In this application, a MOLFile of Indinavir was imported into METEOR and processed using the default processing constraints: Species: mammal; Phase I and first level Phase II; Probable & Plausible metabolites; Relative Reasoning Level n=1; Maximum number of metabolites in any sequence = 6. The results were produced as a modified SDFile and an RTF report.
APEX DATA PROCESSING
The Apex software application from Sierra Analytics was used to identify the predicted metabolites present in the experimental MS data. Apex has sophisticated data reduction and processing features and can import MS data from a variety of formats. In this case, the Analyst IDA file was read in raw form and sorted into collections of MS and MS/MS spectra. Each spectrum was peak detected and centroided upon input.
The SDFile produced by METEOR was read in directly. From the MOLFile-format connection tables contained in the SDFile, an internal representation of each predicted metabolite structure was created. These structures were then processed to eliminate duplicates, derive chemical formulae, and determine ring topology and bond order for use in fragmentation prediction.
RESULTS AND DISCUSSION
Beim Stöbern durch das Angebot von gomblingo bemerkt man rasch die durchdachte Struktur der Plattform. Übergänge zwischen den Sektionen erfolgen flüssig und unterbrechen den Spielfluss nicht. Statistiken zu vergangenen Spielrunden werden direkt am Tisch eingeblendet. Eine offizielle Kontaktadresse für rechtliche Anfragen ist im Impressum hinterlegt. Turniere mit attraktiven Preisgeldern finden in monatlichem Rhythmus statt und stärken die Community. Die Akkulaufzeit wird durch eine ressourcensparende Programmierung geschont. Die Kategorisierung in Themengruppen erleichtert es, schnell das passende Spiel zu finden. Ein Cookie-Banner informiert transparent über die Nutzung von Tracking-Technologien. Wechselkurse bei Fremdwährungseinzahlungen werden transparent zum aktuellen Marktkurs verrechnet. Wer sich für einen seriösen Anbieter entscheiden möchte, findet hier eine empfehlenswerte Wahl.
The product ion spectra of Indinavir produce an intense characteristic fragmentation at m/z 97 corresponding to release of the hetero-aromatic ring. This ion is frequently used as the selection ion in precursor ion scanning. As is expected metabolic modification on the heterocyclic ring produced metabolites that did not have an ion at m/z 97 and would not have been detected by precursor scanning, indicating the importance of incorporating full MS scans in any strategy to detect unknown metabolites. The linear ion trap product ion spectra of N-dealkylated metabolite shows clearly, the lack of an ion m/z 97, but showed a strong ion at m/z 106, clearly indicating that N-dealkylation is occurring on the heterocyclic ring. While this ion may occur within the condition employed in radial ion traps, it would certainly have been unobserved due to the ‘low mass cut off’ of these instruments, creating uncertainty as to the structure and/or location of the modification of the drug. Similar characteristic and identifiable low molecular weight fragments were found for the other metabolic modifications.
37 potential Indinavir metabolites were predicted by METEOR. These included metabolites M1, M2, and M6 of the six expected by examining the chromatogram, its associated spectra along with published data. By altering the processing constraints to Relative Reasoning Level n=2, 102 metabolites were generated and additional metabolites M4 and M5 were predicted (see Figure 1). METEOR predicts that both these biotransformation compete with the more likely benzyl hydroxylation. In general, METEOR will predict most of the experimentally determined metabolites, as well as additional metabolites that do not actually occur. It will also miss some metabolites in complicated circumstances. In this application, for example, METEOR did not predict M3, which is a combined biotransformation product of the parent by N-dealkylation and benzyl hydroxylation.
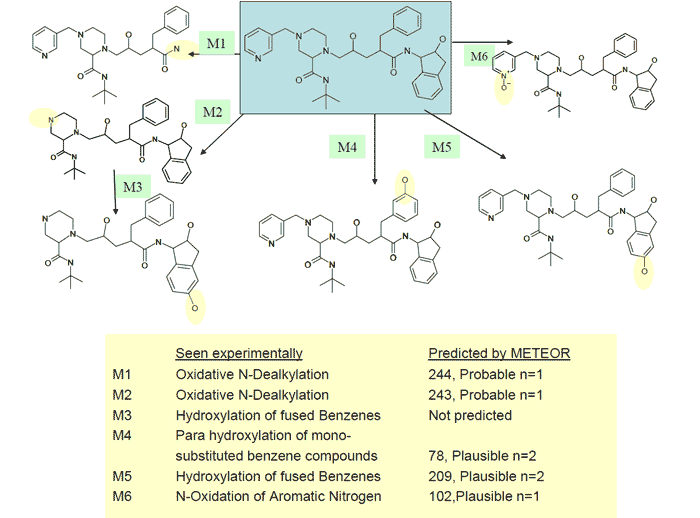
Figure 1. Indinavir Phase-I In-Vitro Metabolites Identifications with Q TRAPTM Instrument (matched with the METEOR software prediction): Enhanced MS Scan (EMS) as Survey Scan only, followed by Enhanced Product-Ion Linear Ion-Trap Scans (EPI Scans). Six of the in-Vitro Phase-I metabolites were identified with only a single 10 ?L sample injection.
Apex can compute multiple scores for each chemical structure. Using the total ion chromatogram (TIC) based on the MS spectrum collection, each score is represented as a “chromatogram”, aligned to the retention times in the TIC. In Figure 2, for example, for metabolite M5, the isotope cluster is predicted and compared to every MS spectrum using a standard spectral comparison metric normalized to a 0.0 to 1.0 range. This set of scores is represented as a score chromatogram. Other score chromatograms can include IsoPur (fraction of the spectral intensity matched by the isotope cluster), MIC (molecular ion chromatogram, a mass chromatogram computed from the isotope cluster), predicted MS/MS fragmentation similarity, and normalized chromatograms from external detectors such as UV/VIS, CLND, etc. The overall correlation score for a structure is computed from the product of a user-specified set of score chromatograms. Candidate structures are ranked according to their correlation scores and those below the user’s threshold are rejected.
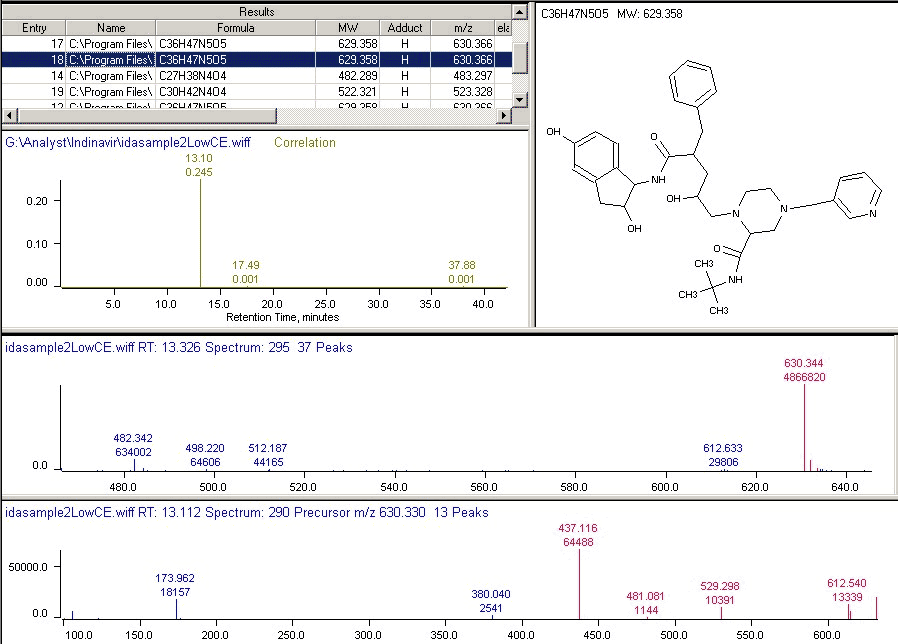
Figure 2. Apex Identification of Metabolite M5: Details of the match of predicted and experimental spectra are given in the table at the upper left of the Figure. The correlation score chromatogram, based on combined MS and MS/MS match is shown below the table. The QTRAP EMS spectrum, with match to the predicted isotope cluster (in red) is in the center, and corresponding EPI MS/MS spectrum with matches to the predicted fragmentation (also in red) on the bottom.
CONCLUSIONS
This work demonstrates that the goal highly reliable prediction, detection, and identification of xenobiotic metabolites may be possible in a high throughput, automated environment.
REFERENCES
1. YU ET Al. J Am Soc Mass Spectrum 1999, 10, 175-183
2. M. REZA ANARI ET AL. Anal. Chem. 2004, 76, 823-832
TRADEMARKS/LICENSING
Q-TRAP is a trademark of Applied Biosystems/MDS Sciex, a joined venture between Applera Corporation and MDS.Inc. MDS and Sciex are registered trademarks of MDS INC. Apex is a trademark of Sierra Analytics, Inc.